Unleashing the Potential of Human Tissue Biomarker Analysis with Spatial Epigenetic Profiling
Dr Michael Eyres, Lead Scientist at MDC, has been working on applying MDCs biomarkers expertise to a novel approach in drug discovery.
July 2024

We are unleashing the potential of human tissue analysis with spatial epigenetic profiling within our biomarkers capability.
Biomarkers improve our understanding of drugs, disease, and drug-target interactions within patients and are key to unlocking a better understanding of translating drugs from the pre-clinical into a clinical setting.
Answering key clinical questions covered in our blog ‘What is a biomarker strategy, and why is it important?’ will mean greater chances of success in drug discovery.
Medicines Discovery Catapult (MDC) has a suite of advanced technologies for biomarker discovery and development, and we are now introducing the newest technology that has been validated by the team at MDC.
Dr Michael Eyres, Lead Scientist at MDC, has been working on applying MDCs biomarkers expertise to a novel approach in drug discovery. Utilising MDC’s expertise in spatial proteomic and transcriptomic techniques on a wide range of tissues and diseases, this latest approach harnesses the forefront of spatial profiling technology to enable genome-wide epigenetic profiling at near-single-cell resolution in frozen tissue. In this blog, Michael takes us through his findings with epigenetic profiling at MDC.
Why Spatial Analysis?
While bulk analysis can be useful for exploring global changes, spatial information is essential to understanding drug responses and disease mechanisms within areas of tissue samples. Spatial analysis in human tissue can be retained for a range of biomarkers (proteins, transcriptome, lipids, metabolites) and examined using the innovative platforms and techniques available at MDC. The most recent advancement in this area is spatial epigenetic profiling, which measures the structure of DNA and how it influences the expression of genes.
What is epigenetic profiling?
Each of the nuclei in our cells contains approximately 2 meters of tightly packed DNA. This DNA is wrapped around nucleosomes, which form the fundamental subunits of chromatin. How tightly packed these nucleosomes are can influence the accessibility of transcription factors and other regulators of gene activity, causing the expression of a particular gene to increase or decrease. Any change in gene expression occurring independently of DNA mutations is referred to as epigenetics, and epigenetic changes are tightly linked to ageing and most diseases.
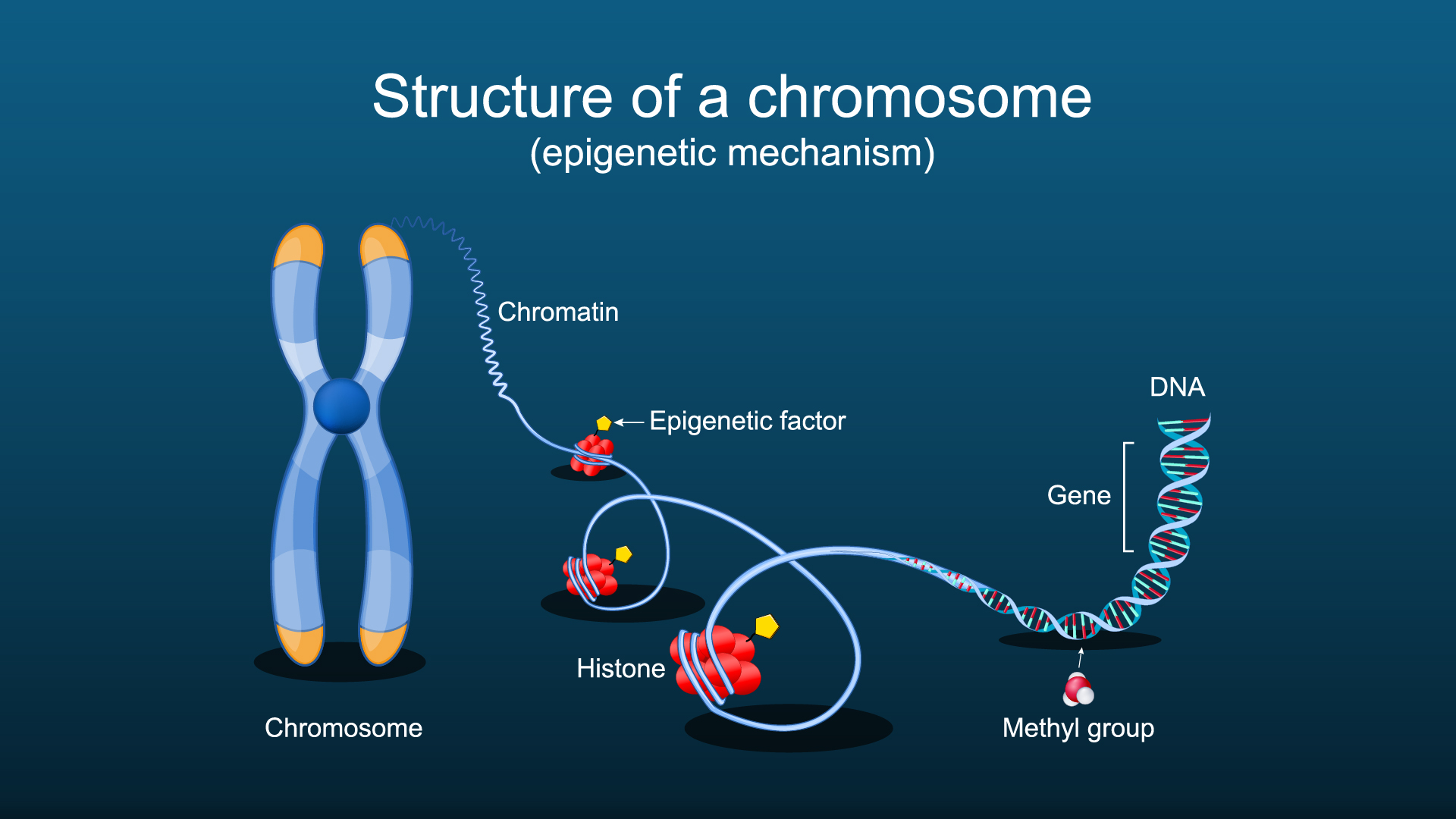
New Technology Enabling Frozen Tissue Analysis at Near Single-Cell Resolution
Assays for Transposase-Accessible Chromatin Sequencing, or ATAC-Seq, is a method to investigate the accessibility of DNA which utilises the Tn5. This transposase can simultaneously cut DNA and integrate a loaded DNA barcode (such as a sequencing adapter). As open chromatin is more accessible, more Tn5 integrations occur in those regions, and by sequencing the barcodes, the accessibility of chromatin genome-wide can be measured.
Traditionally, ATAC-seq has only been possible in bulk tissues or single cells. However, AtlasXomics have developed a microfluids device (Flowgel) that enables it to be used in frozen tissues at near-single-cell resolution. The chips contain multiple channels, with each channel carrying a different set of barcodes. By running two chips perpendicular to each other, the exact XY coordinates can be deconvolved after sequencing by looking at the combination of barcodes present in each read.
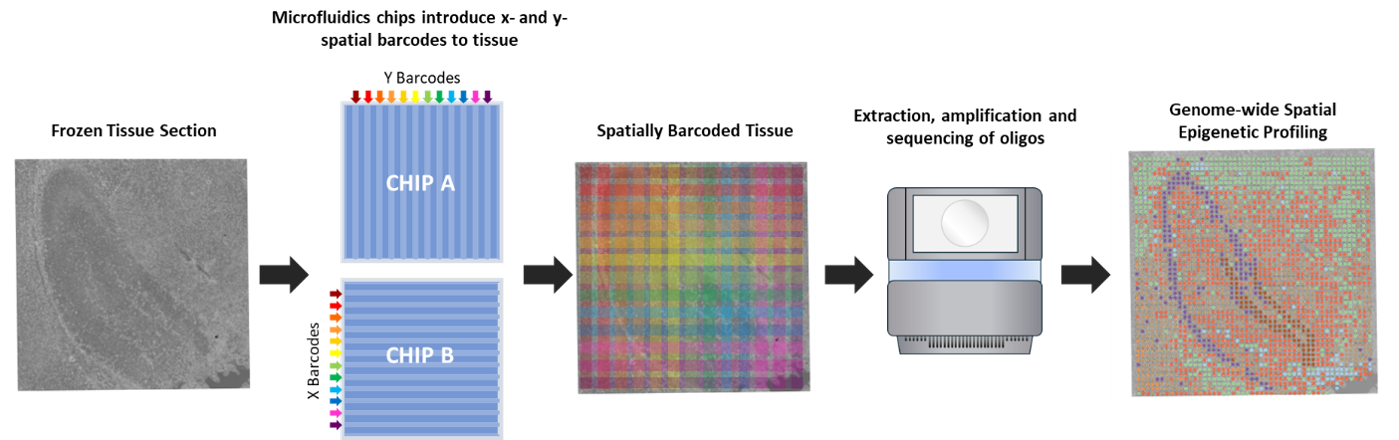
Spatial epigenetic profiling of frozen tissue: frozen tissues are fixed, permeabilised, and tagmented. The Tn5 transposase cuts open regions of chromatin, and microfluidics chips are used to introduce unique barcodes along the channels in the X and Y directions across the tissue. After extraction, amplification, and sequencing of DNA, the XY coordinates of each read can by spatially mapped based on the combination of X and Y barcodes. This enables genome-wide, spatial profiling of chromatin and near single-cell level.
Flowgel has been extensively tested and validated at MDC in various types of tissues and diseases.
- Flowgel 96 – 3.8 x 3.8mm area assayed at 20um resolution.
- Flowgel 210 – 5.2 x 5.2mm area assayed at 10 or 15um resolution.
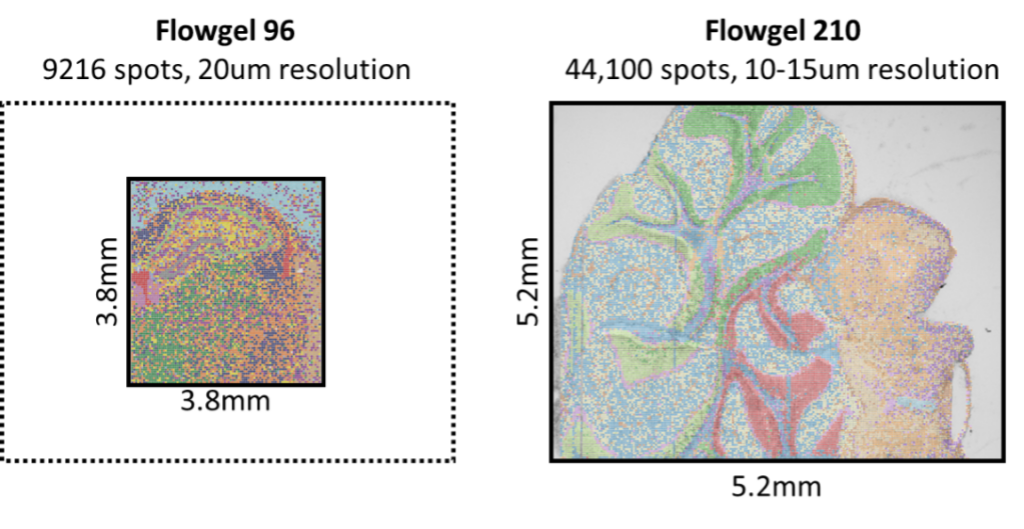
Size and Resolution
Depending on the chips used, spatial profiling can either be carried out in a 3.8 x 3.8 region with 20um resolution, or 5.2 x 5.2mm region with 10-15um resolution.
Using the AtlasXomics Microfluids Device ‘Flowgel’ at MDC
Case Study 1: Epigenetic Profiling of Ageing in the Brain
Spatial ATAC-seq was carried out on the hippocampus regions of a young and an aged brain. Various epigenetic clusters were identified which corresponded with the underlying morphology of the tissues, e.g., cluster 6 corresponded to the CA1-4 region. Other clusters could be identified due to the accessibility of genes associated with cell type markers, such as Myelin Basic Protein (MBP), a marker of oligodendrocytes, being highly accessible in cluster 2. Differential gene accessibility analysis was carried out to identify genes with altered accessibility with ageing, highlighting hundreds of genes, many of which with known roles in ageing.
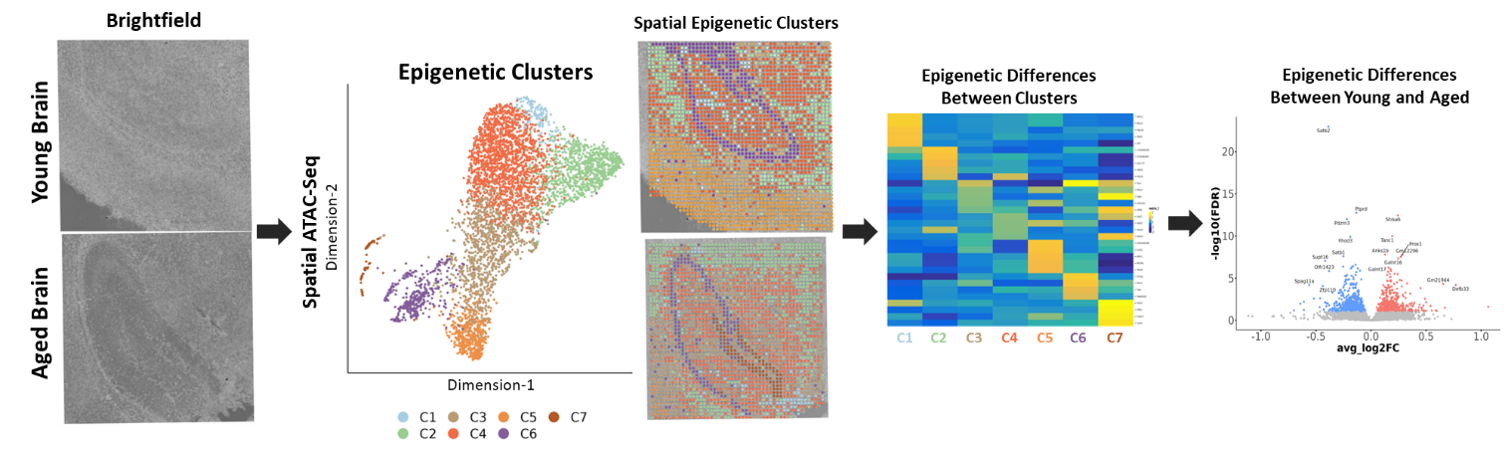
Epigenetic profiling of young and aged brains:
Spatial ATAC-seq was carried out on hippocampus regions from young and aged brains. Genome-wide profiling of open chromatin identified seven distinct clusters with accessible genes in known cell type markers. Differentially accessible gene analysis between young and old brains identified epigenetic changes in hundreds of genes, many of which have previously been linked to ageing.
Case study 2: Loss of TF Binding in Alveolar Macrophages During Lung Fibrosis
Idiopathic Pulmonary Fibrosis (IPF) is a highly aggressive lung fibrosis disease with no known cause. Spatial ATAC-seq was carried out on biopsy samples from a donor with IPF and a healthy control. One cluster was highly epigenetic distinct from the others and was associated with alveolar macrophage genes such as CD11C. Differential motif accessibility was carried out, which looks at the accessibility of transcription factor binding sites across the genome. This identified a transcription factor with binding sites that were highly accessible in healthy alveolar macrophages. In IPF samples, however, these binding sites were much less accessible, suggesting that the transcription factor is less active.
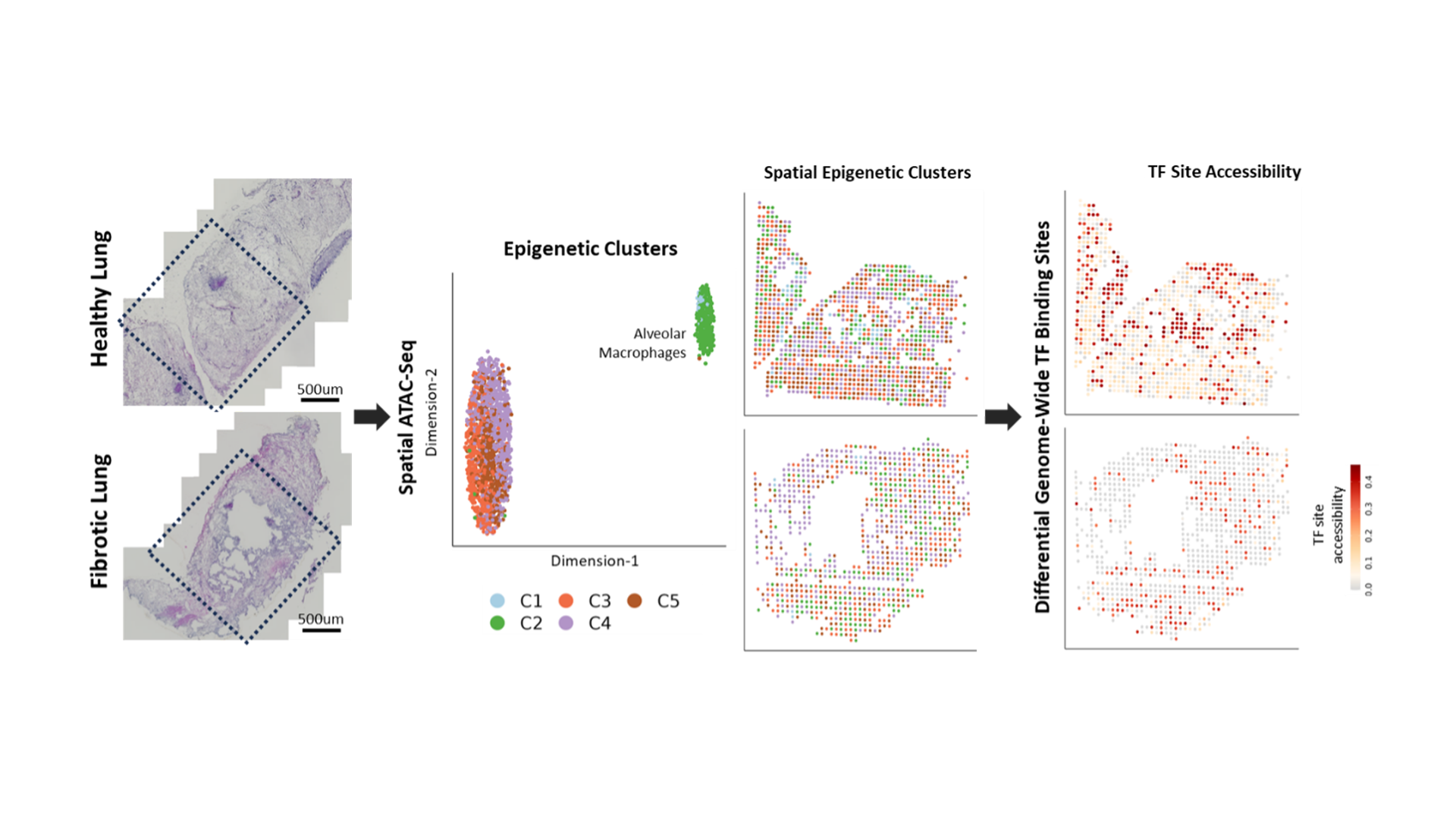
Spatial ATAC-seq of healthy and fibrotic lungs: Spatial ATAC-seq was carried out on lung tissue from a donor with Idiopathic Pulmonary Fibrosis (IPF) and a healthy control. One of the clusters was epigenetically distinct from the others and was enriched for alveolar macrophage markers. Differential motif accessibility analysis showed that binding sites for the transcription factors were less accessible across the genome in IPF macrophages.