Disentangling cellular pathology and therapeutic action with live-cell imaging
July 2026
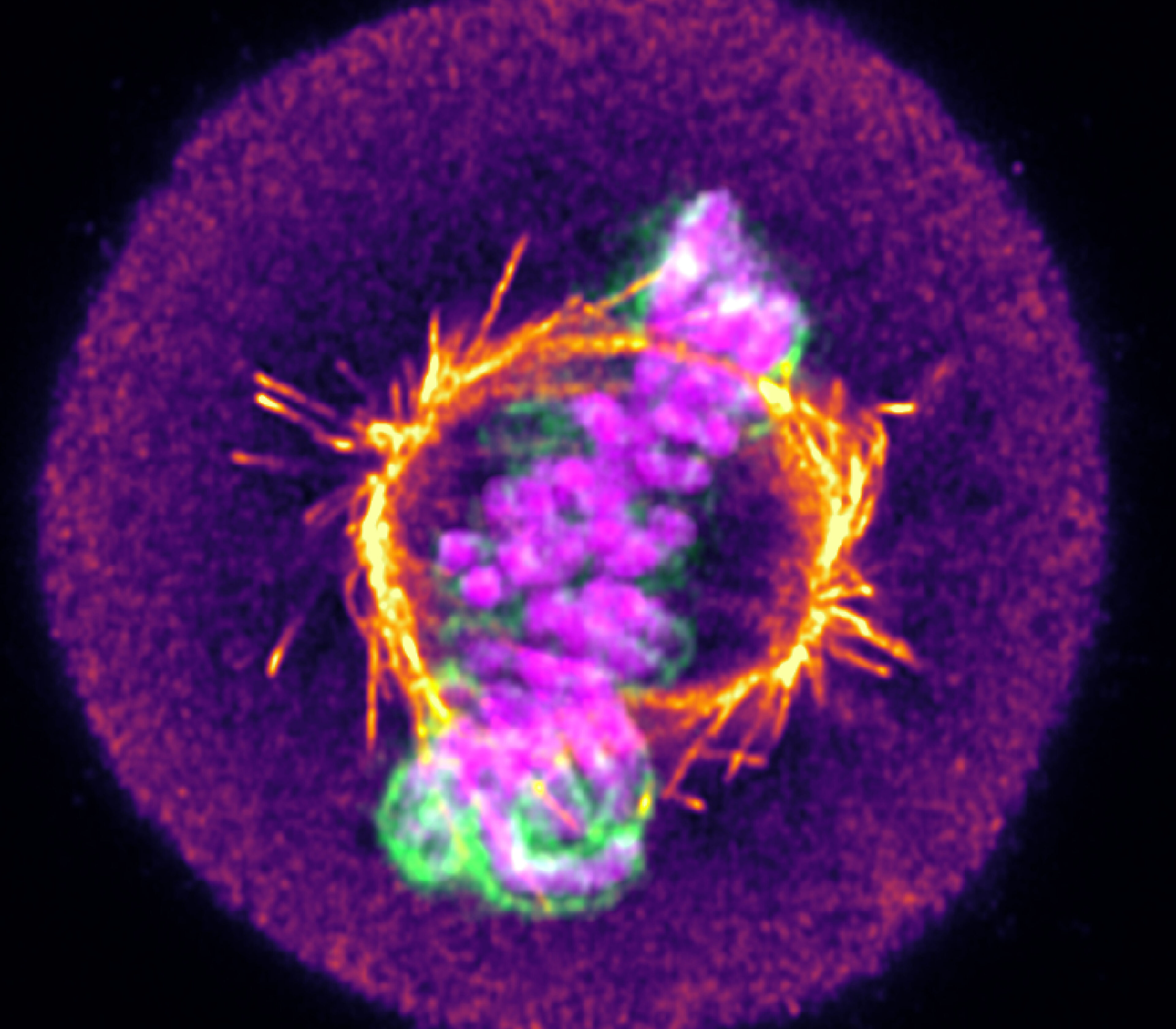
Medicines Discovery Catapult has invested heavily in live-cell imaging technology and expertise because of its unique capacity for disentangling pleiotropic phenotypes. Below, we discuss why it’s an essential asset and provide three examples of how live-cell imaging can elucidate pharmacological and cellular dynamics that remain hidden at fixed endpoints.
Methodological bias is a frequent pitfall in biological imaging, where conclusions are skewed by the way a sample is prepared or visualised. For example, fixation with paraformaldehyde can either enhance or diminish putative liquid-liquid phase separation, where intrinsically disordered proteins condensate to form membrane-less organelles. Similarly, fixation was found to exclude transcription factors from mitotic chromatin, obscuring a mechanism of transcriptional inheritance called mitotic bookmarking. This chemically induced distortion is not alone; snapshots of dense subcellular environments at low resolution can make molecules appear bound to structures when no true interaction exists. Fortunately, avoiding these artefacts is conceptually simple: validate fixed endpoints with live-cell imaging to understand the dynamic continuum of molecular and morphological changes that link two phenotypic states.
All work was conducted using a 63X 1.4NA objective lens on a Zeiss LSM880 laser scanning confocal microscope equipped with an Airyscan module.
In contrast to the textbook presentation of static oval shaped organelles, mitochondria form an interconnected and highly dynamic network capable of migrating throughout the cell and undergoing fusion and fission. Fission facilitates the detachment of damaged mitochondria for autophagy and clearance, while fusion coordinates network functionality, particularly in response to metabolic stress. Dysregulation of the tightly controlled fusion-fission balance can impact a cell’s metabolic, proliferative and apoptotic state, and is a hallmark of Alzheimer’s disease, linked to cardiomyopathy, and might even drive a metastatic cancer phenotype. The picture is further complicated by the discovery of fission signatures with distinct positional and molecular properties that govern the fate of separating mitochondria. Because of this complexity and potential for phenotypic redundancy, for example, where a reduction in fusion could appear as an increase in fission, live cell imaging is necessary to study these processes. To this end, we established high spatiotemporal imaging approaches to visualise mitochondrial dynamics in cultured cells using NAO and Mitotracker Green dyes, respectively (Figure 1a,b). Fusion and fission events could be identified and tracked in both instances, enabling us to map kinetic trajectories and evaluate how specific pathological conditions or therapeutic compounds alter network equilibrium.
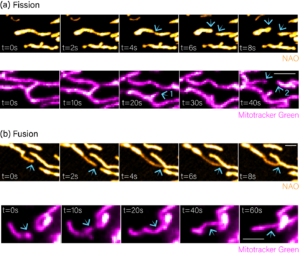
Figure 1: Visualising mitochondrial fission and fusion. (a) Movie stills showing fission events visualised using NAO and Mitotracker green dyes, respectively. (b) Movie stills showing fusion events visualised using NAO and Mitotracker green dyes, respectively. Blue arrows show the position of fission or fusion.
Mitotic cell division is a stepwise process responsible for multiplying a single zygote into the 30 trillion cells that form an adult human. In mitosis, replicated DNA in the form of condensed chromosomes is aligned at the cell equator forming the metaphase plate then equally segregated into two daughter cells during anaphase (Figure 2a). Segregation is physically driven by the attachment of microtubule fibres to sister chromatids, which depolymerise and pull chromatids to opposite cellular poles. Attachment errors cause asymmetric segregation that results in daughter cells with an abnormal number of chromosomes, a state called aneuploidy. To combat this, cells have evolved mechanisms to sense attachment errors and correct these as mitosis progresses. Therefore, the attachment process can be thought of as an ongoing battle between the formation of improper attachments and their correction. Normally the efficiency of correction vastly outweighs the rate of erroneous attachment formation. However, in many cancers the balance is shifted, resulting in the regular generation of aneuploid cells that drive malignant evolution and therapy resistance. Due to the dynamic nature of mitosis and attachment correction, this must be studied using live cell imaging.
To investigate mitotic errors generated through a failure of attachment correction, HeLa cells were stained with the far-red dye SiR-DNA and imaged every three minutes for twelve hours (Figure 2). Analysis of mitotic progression showed three phenotypes, (1) anaphase where the two chromatin masses clearly separate (Figure 2a), (2) anaphase with lagging chromosomes that resolve (Figure 2b, lazy chromosome), and (3), anaphase with lagging chromosomes that persist (Figure 2c, lagging chromosome). Resolution of chromosome lagging demonstrates the success of attachment correction, while persistence shows its failure and the generation of aneuploidy. Crucially, in early anaphase these processes appear falsely equivalent, and would be considered identical in a fixed endpoint setting despite having phenotypically opposite outcomes (Figure 2b,c).
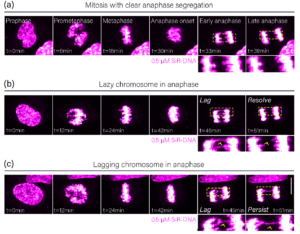
Figure 2: Analysis of anaphase phenotypes. Movie stills showing HeLa cells progressing through mitosis with no lagging in anaphase (a), a lazy chromosome in anaphase (b), and a lagging chromosome in anaphase (c). Yellow boxes show zoomed regions. Yellow arrows indicate the lazy and lagging chromosomes, respectively.
RNA therapeutics represent one of the most promising new drug modalities by providing an approach to therapeutically modulate previously intractable or “undruggable” targets. This promise is, however, coincidental with delivery challenges due to the instability of RNA in serum and the inability of RNA to freely translocate the plasma membrane. A widespread solution is the encapsulation of RNA in a delivery vehicle that protects it from damaging biological interactions and delivers it to the cytosol of target cells. The latter requires that the vehicle-payload complex internalises into endocytic vesicles then escapes through rupture of the limiting membrane. Vehicle chemistry is optimised to induce rupture at a specific step in the endocytic pathway, therefore, understanding vehicle-payload complex localisation to endosomal subcompartments is necessary for therapeutic evaluation. Visualising the localisation of labelled vehicle-payload complexes to individual endosomes using fluorescence microscopy is confounded by vesicle size, which is often below the resolution limit. Even when using Airyscan super-resolution imaging of fixed cells treated with labelled vehicle-payload complexes and stained with antibodies against EEA1 (early endosomes) or Rab7a (late endosomes), interpretation is muddled (Figure 3a,b). This is due to the density of the endocytic network, where overlapping fluorescence from spatially distinct vesicles obscures the precise localisation of therapeutics (Figure 3a,b). Live-cell imaging can address this by visualising the co-dynamic relationship between vehicle payload complexes and endosomes, which is based on the rationale that if two colocalised signals migrate concordantly over multiple time points, it is reasonable to conclude they are associated. To demonstrate this, we used CRISPR genome engineering to label EEA1 and Rab7a with the fluorophore mStayGold, then incubated these cells with vehicle-payload complexes labelled with Alexa Fluor 647. Live cell imaging showed these complexes rapidly transit the EEA1 compartment, demonstrating the power of this approach to identify transient states (Figure 3c). Furthermore, we found that vehicle-payload complexes enter Rab7a positive vesicles en route to degradation, as evidenced by the persistent spatiotemporal colocalisation of complex and Rab7a signals (Figure 3d,e). Ultimately, this dynamic tracking platform enables the intracellular fate of RNA delivery vehicles to be mapped with confidence, providing a robust method to evaluate how changes in vehicle chemistry can improve cytosolic payload delivery.
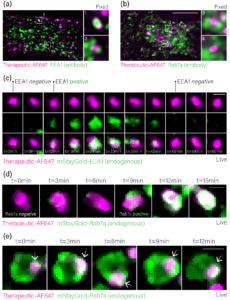
Figure 3: Analysis of RNA therapeutic localisation to endosomes. (a) Airyscan image of a HeLa cell treated with Alex Fluor 647-labelled vehicle-payload complexes, fixed, and stained with an antibody against EEA1. (b) Airyscan image of a HeLa cell treated with Alex Fluor 647-labelled vehicle-payload complexes, fixed, and stained with an antibody against Rab7. (c) Movie stills of Alex Fluor 647-labelled vehicle-payload complexes transiting the EEA1 compartment in live HeLa cells. (d) Movie stills of Alex Fluor 647-labelled vehicle-payload complexes entering the Rab7a compartment in live cells. (e) Movie stills of Alex Fluor 647-labelled vehicle-payload complexes demonstrating persistent spatiotemporal colocalisation with a Rab7a positive endosome.
Static endpoints only tell half the story. Talk to our team about how live-cell imaging could de-risk and sharpen your programme.



Xenium at MDC Case Study: Hypoxia in Head and Neck Cancer
By looking beyond the diffraction limit, we have established a method to quantitatively distinguish low-grade breast cancers that appear identical by traditional means. Ultimately, this nanoscale insight could expand access to novel HER2-targeted therapies by reliably identifying low expressing cohorts that would benefit from intervention.
Lipid nanoparticles (LNPs) are recognised as a leading delivery platform following clinical success in vaccine and therapeutic applications. Medicines Discovery Catapult (MDC) has built a preclinical platform to evaluate LNPs against two major challenges in the field. In this blog, Lead Scientist, Dr Phil Auckland, presents a case study of this workflow using intravenously administered LNPs encapsulating mRNA, which differentially target four major liver cell types.
