Medicines Discovery Catapult de-risks new medicine discovery, provides access to technologies, skills, and knowledge, and runs impactful R&D partnerships.
We connect innovators to a broad and diverse support network for the next step of their journey. We provide insights, critical commentary, and guidance for the sector and the development of new policy.
We are part of the Innovate UK Catapult Network, co-funded by industry and government. We are not-for-profit and commercially driven. We reinvest time, money, resources and discoveries back into Life Sciences to help our sector thrive.
Find out moreWe have supported 397 innovators, helping them raise over £1.9bn of R&D investment and supporting them to take five new drugs to clinical trials.
Our impactSME funding secured post-engagement
grant income raised in collaboration
compounds supported into the clinic
partner products progressed to market
assays supporting clinical development
organisations supported

We de-risk new medicine discovery and provide access to technologies, skills, and knowledge.
Our experts apply the very best new laboratory technology to validate ideas. We help build long-term success to reshape drug discovery.
We use our expertise and networks to design robust R&D roadmaps for the quickest, safest route to patients and commercial success.
We use our global connections to create momentum for innovators so they can access broader support, secure investment, and get to market.
Explore more


We tackle global challenges in Life Sciences by stimulating innovation and addressing unmet patient need through our national programmes.
We connect patient communities, charities, global biotechs, academia, and pharma to accelerate discoveries and improve lives.
Find out moreRead our stories showcasing how we have helped drug discovery SMEs and innovators.
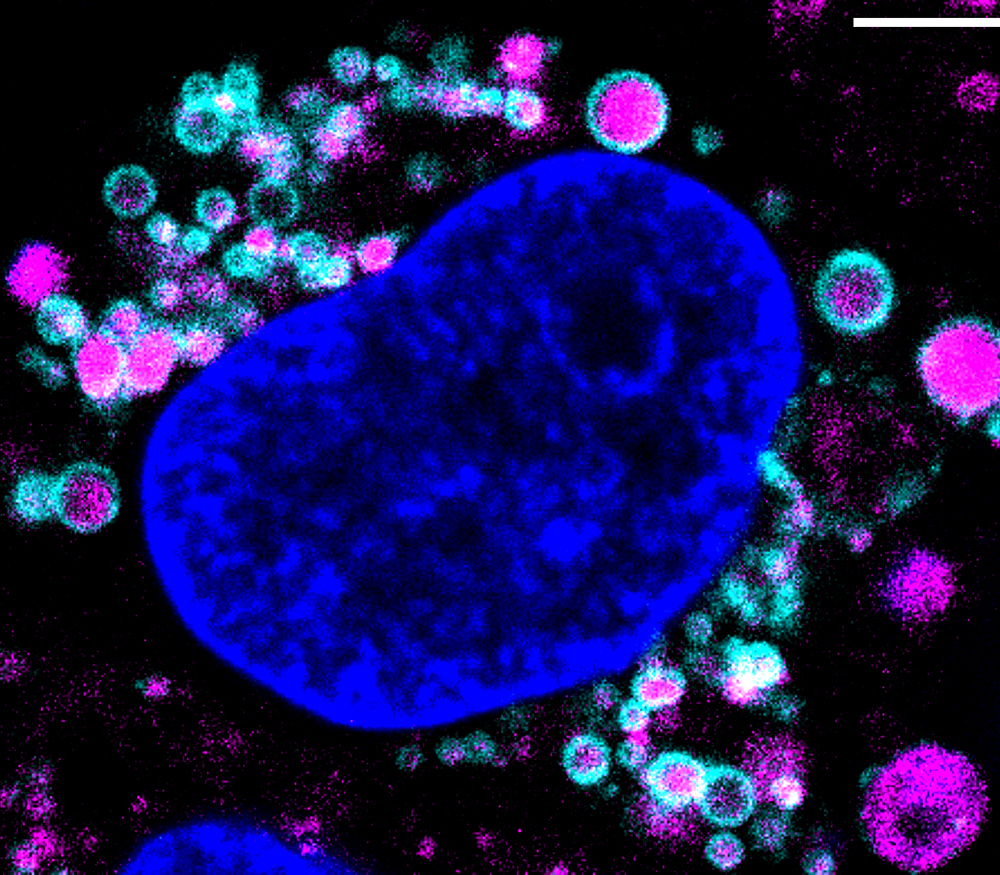

We can support you with our R&D consultancy, Collaborative R&D services, and grant support.
Learn moreNews, blogs, events, reports and more. Our latest press releases, media appearances, events and news commentary.
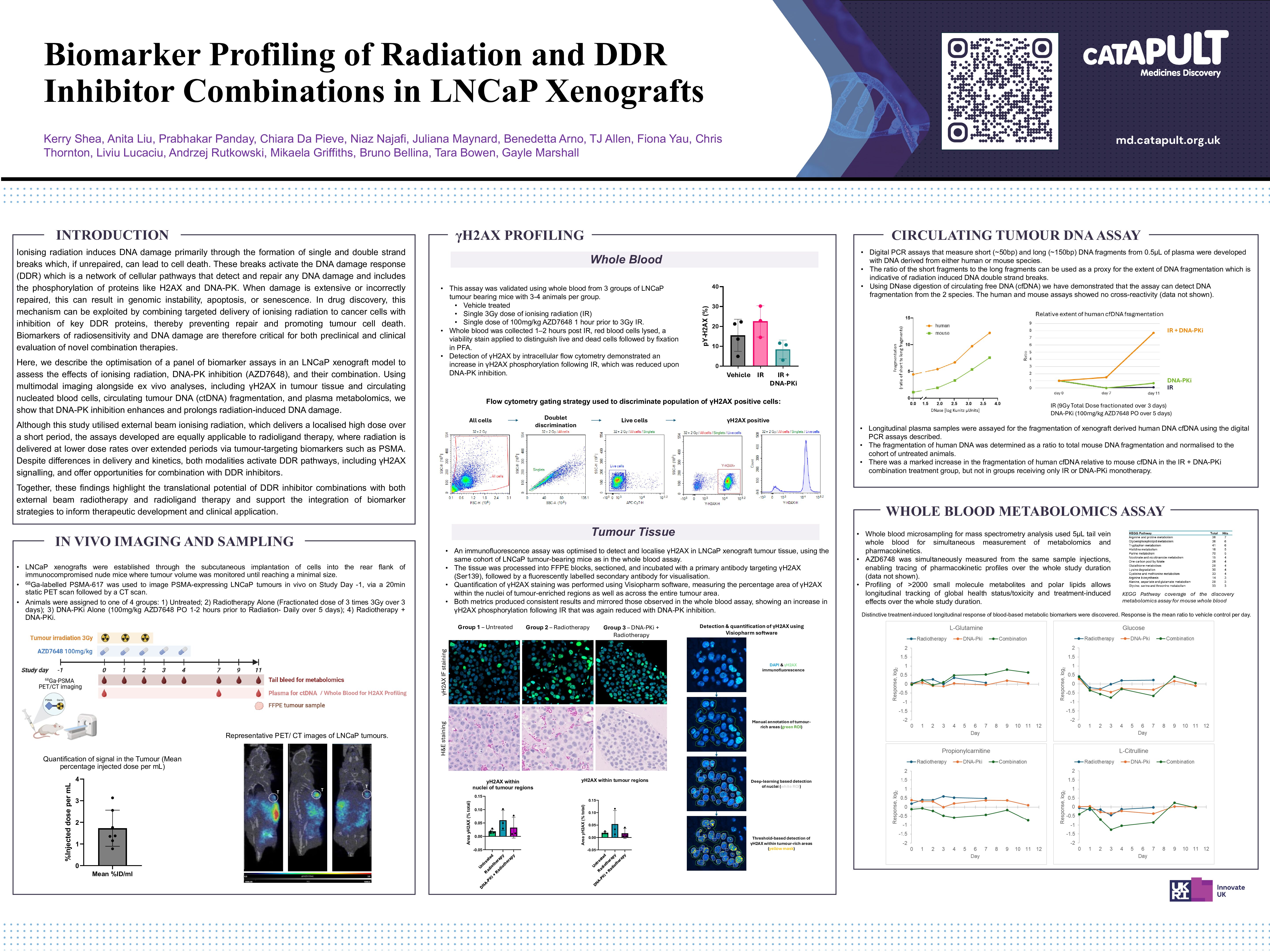















Get the latest news, blogs, reports, publications and upcoming events direct to your inbox every month.
See how we can help support you with your next drug discovery project.
Speak to us