MedChemica is an in-silico drug design AI company with a suite of software and databases for drug design – they also support research projects such as the COVID moonshot project, alongside antibacterial and oncology projects.
Why use in-silico drug design?
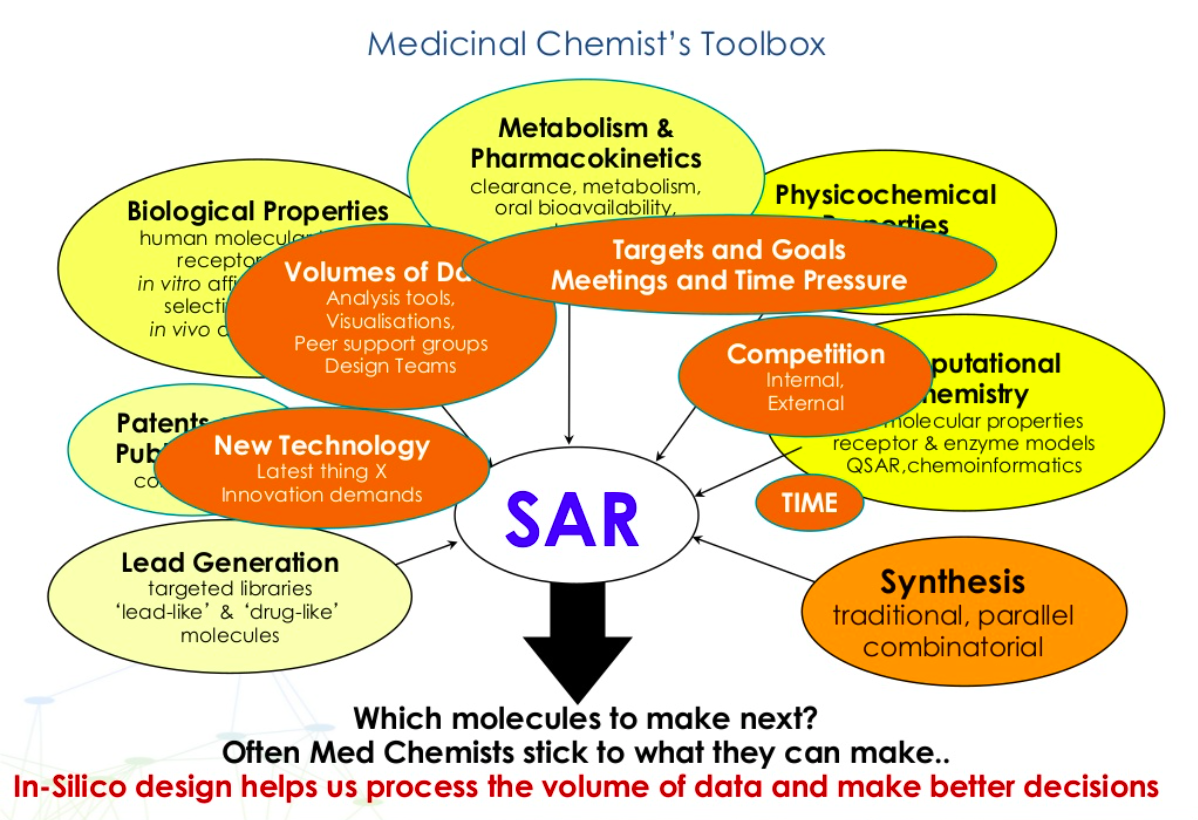
In-silico techniques help analyse the data available unbiasedly, refine compound design and achieve the outcomes for a good quality candidate drug in fewer iterations.
2D computational methods assist in processing the vast amount of data available in an unbiased way to support better decisions on which molecules to progress and allows ranking of generated ideas.
What to do
It is key to obtain as much quality information on the compound as possible from various sources i.e. SAR from literature, patents, and structures, and use mathematical models to automate structure-activity decisions such as Free Wilson analysis. This determines the contributions each of the substituents or structural elements make to the activity/potency of the parent molecule. A more sophisticated, automated method is permutative matched molecular pair analysis (MMPA) which compares the relationships and properties of 2 very similar molecules.
What to watch out for
2D plots can provide detailed information but care should be taken to use the same scale on the x/y axis and that data are measured on a continuous scale. Limitations of 2D models should be considered – descriptions, atoms, or group patterns are used to encode molecules, models are only as good as the dataset from which they are generated, and every prediction is liable to errors.
3D design/structure-based drug design can be based on single molecule structures, conformational and torsional analysis, protein/ligand structures, docking and scoring and force field/scoring. 3D design can include free energy perturbation, quantum mechanics, and molecular dynamics.
Structural modifications, size and lipophilicity are all important in compound development. The lipophilicity of a molecule can be estimated using calculated lipophilicity (ClogP). This should fall between 1-3 on the LogD/P scale, too polar or too lipophilic can generate problems with the compound.
Key to a solid understanding of the area are literature references. MedChemica provides a library of references.
In addition, the company currently has a rare opportunity with a community project searching for SARS COV-2 inhibitors of one of the proteases. The project can be followed on Twitter @covid_moonshot.
This article is based on Al’s talk from the MDC Connects webinar series. Watch the session Al took part in –Optimising the Compound:

About the author

In 2012 Dr Al Dossetter co-founded MedChemica Limited centred around the technology of Matched Molecular Pair Analysis (MMPA) as a method of accelerating medicinal chemistry. Previous to MedChemica Al gained his PhD from Nottingham University and after post-doctoral research at Harvard University joined AstraZeneca (AZ). He spent 13 years in medicinal chemistry spread across oncology (hormonal and kinase inhibitors), inflammation (OA and RA, enzyme inhibitors and GPCR targets) and diabetes (obesity, GPCR and enzyme inhibitors), delivering multiple projects and candidate drugs.





















 Homepage image")