Proteins are the targets for most marketed drugs today, and high-quality purified proteins are required for the drug development process. Proteins are also important biological therapeutics, so the ability to produce high quality proteins is essential.
What is involved in protein production?
Highly expressed proteins can often be readily obtained from natural sources; however, proteins that are only produced in small quantities within cells are required to be generated recombinantly. This often involves making a synthetic, codon-optimised gene for the protein of interest, cloning it into a plasmid expression vector and then transfecting into cultured cells to express the recombinant protein. Common expression systems include E.coli, baculovirus/insect cells or mammalian cells, each with their advantages and disadvantages.
Success in obtaining the required protein often requires careful engineering of the protein constructs to be used, for example, is the full-length protein required or only a particular catalytic/functional domain, are post-translational modifications important for function/stability etc. Such considerations are also used to select the right expression system for the target protein in question. Addition of affinity tags (e.g. His6, Flag) can simplify purification whilst use of protein fusion partners (e.g. MBP, SUMO) can increase protein solubility.
Protein purification is then normally performed using a range of liquid chromatography technologies including affinity chromatography, ion exchange chromatography and size exclusion chromatography.
It is very important to monitor and determine the quality of the protein being produced and this is done using a variety of methods. SDS-PAGE gels give an indication of purity and approximate molecular size, absorbance at 280nm estimates protein concentration, analytical size exclusion chromatography provides the molecular size of the protein or complex in solution. Mass spectrometry gives an accurate molecular mass that can be used to confirm the identity of the purified protein. A functional assay, if available, is useful to determine if the purified protein retains its expected activity.
Protein structure determination by X-ray crystallography
Knowledge of protein structure is important as it informs on protein function and can enable the rational chemical design of drug molecules. One common method used to determine the atomic structure of proteins is X-ray crystallography. This relies on the ability of proteins to form crystals that can diffract X-rays. However, as there is currently no way to predict in advance the optimal conditions under which a particular protein will crystallise, it requires an empirical process where large numbers of different combinations of precipitants, buffers, and salts are incubated together with the protein of interest to identify which conditions will produce suitable crystals. Such crystallisation screening is often carried out by robotic systems in 96-well plates using vapour diffusion methodologies.
Once crystals have been successfully obtained, data is collected by exposing the crystals to intense X-ray beams, often at specialised X-ray synchrotron facilities. The diffraction data typically consists of a large number of images of the X-ray diffraction pattern that are obtained as the crystal is rotated in the X-ray beam.
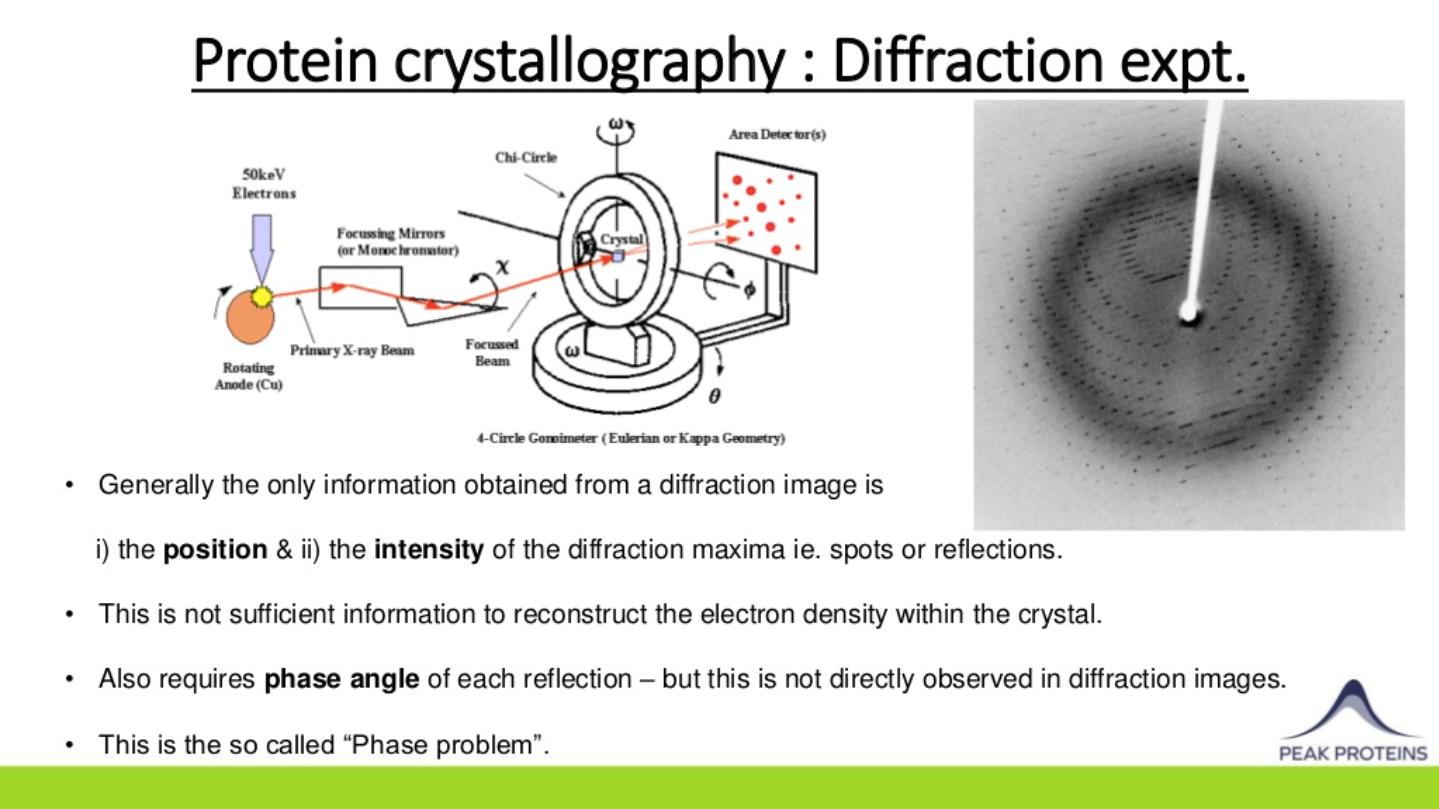
Knowledge of the position and intensity of each diffracted spot or reflection in such images is generally not sufficient to reconstruct the protein’s electron density within a crystal. Information on the relative phase angle of each reflection is also required and this can be determined using a variety of methods. Molecular Replacement relies on the availability of similar/homologous protein structures (sequence ID of >30%) to use as an initial model. If no homologous structures are available, then Multiple Anomalous Dispersion (MAD) methods can be used in which native methionine residues are replaced with selenomethionine during protein expression. The resulting small differences in diffraction intensities are then used to calculate the required phase information.
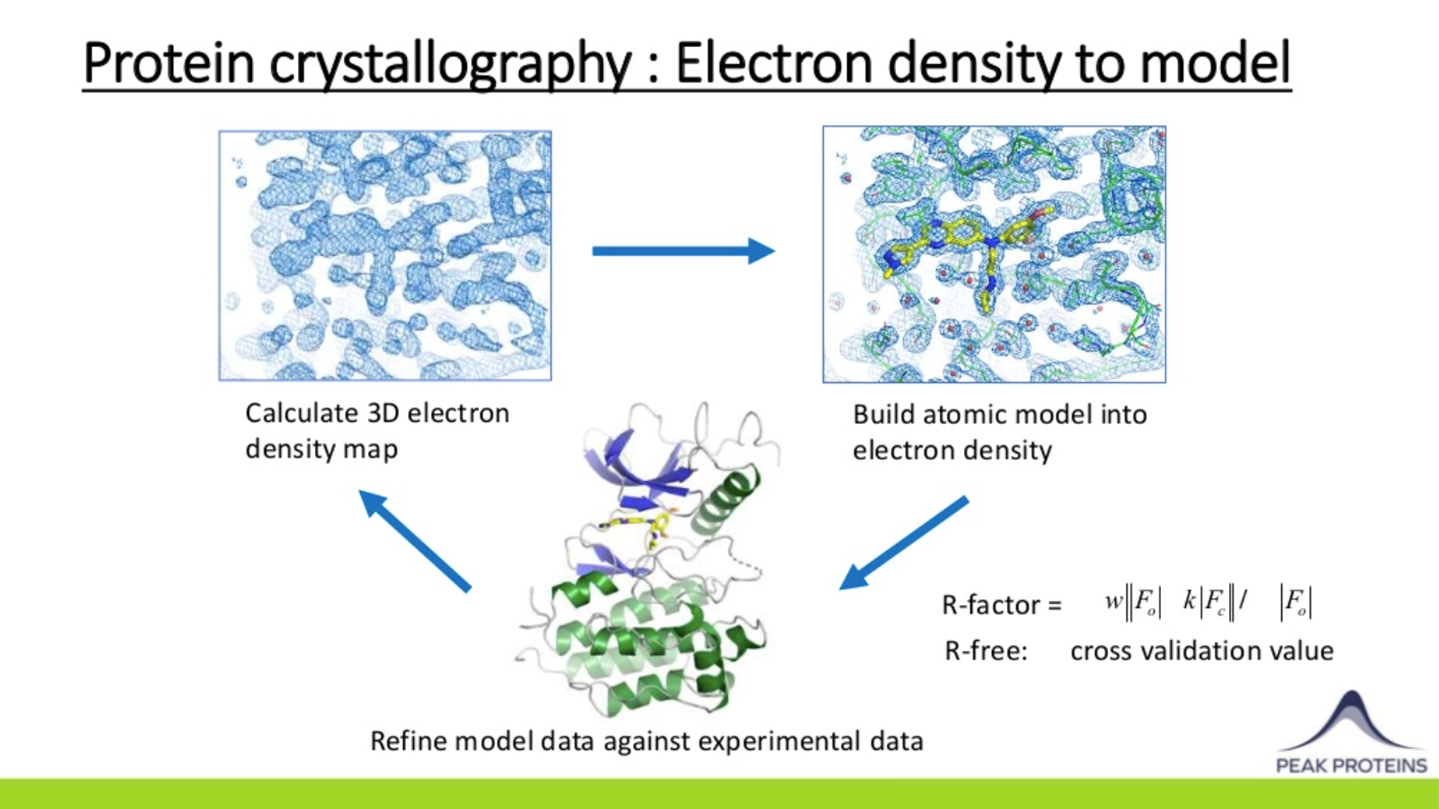
Once initial estimates of the phases are obtained, electron density maps of the protein within the crystal can be calculated and visualised using computer graphics. This allows a model of the protein to be built which can then undergo cycles of refinement against the experimental data to improve the model and provide even better phases until there is convergence.
Just as with purified proteins, it is important to monitor and determine the quality of the protein models that are generated. Protein structures determined by X-ray crystallography should be explained by the experimental data e.g. have low Rfactor & Rfree values. The quality of the experimental data itself should be assessed by monitoring its resolution, completeness, redundancy, signal-to-noise, and merging statistics. Finally, protein models should also make stereochemical sense. This can be judged readily by examining Ramachandran plots to ensure that the amino acid backbone phi/psi angles fall within expected limits.
This article is based on Derek’s talk from the MDC Connects webinar series. Watch the session Derek took part in –Structural Approaches for Drug Discovery:

About the author

Derek Ogg is Chief Scientific Officer at Peak Proteins. He carried out his PhD in Biophysics at the University of Leeds. After postgraduate studies he moved to Sweden to work as a protein X-ray crystallographer with number of biotech and pharma companies including Pharmacia and Biovitrum as well as the Structure Genomics Consortium in Stockholm. On his return to the UK he worked for ten years at AstraZeneca, Alderley Park before joining Peak Proteins as CSO in 2015.



















 Homepage image")